Ion AmpliSeq Designer
Special Instructions are only supported when selecting the GeneStudio Instrument at this time.
Yes. Starting from Genexus software version 6.6 DNA designs created from references other than hg19 are supported for use with the Genexus Instrument.
Yes, please refer to the following table as a general guideline on product support by instrument. For info on any specific panel of your interest, please refer to the available instrument selection ordering options in the Order Preview process that can be launched from each panel page.
| Products: | Instrument | |||
|---|---|---|---|---|
| Genexus(1) | GeneStudio | |||
| AmpliSeq Made-to-Order DNA (All genomes) - up to 275 bp | ✓ (2) | ✓ | ||
| AmpliSeq Made-to-Order DNA (All genomes) - 375 bp | ✓ | |||
| AmpliSeq Made-to-Order RNA (RefSeq) | ✓ (3) | ✓ | ||
| AmpliSeq HD DNA (hg19) | ✓ (2),(4) | ✓ | ||
| AmpliSeq HD RNA (RefSeq) | ✓ (3) | ✓ | ||
| AmpliSeq On-Demand | ✓ | ✓ | ||
| Oncomine tumor specific panels | ✓ | |||
(1) Genexus software 6.8; (2) 1-pool and 2-pool designs only; (3) Gene Fusion designs only; (4) cfDNA/FFPE (75-125 bp) designs only
In general, Community panels for DNA are supported on all sequencing platforms and all other Community panels are only supported on the GeneStudio instrument. However, exceptions apply. For info on the supported journeys for any specific Community panel of your interest, please refer to the available instrument selection ordering options in the Order Preview process that can be launched from each panel page.
In general terms, White Glove designs follow the same support model of AmpliSeq Made-To-Order and AmpliSeq HD designs (see FAQ #4 above). Custom AmpliSeq White Glove methylation panels are only compatible with GeneStudio. Special Instructions and other peculiar features associated to White Glove designs might limit support on one or more instrument (to be assessed case by case at the White Glove submission stage based upon design requirements).
When ordering a Kitted panel, the user is sent to the appropriate product page on thermofisher.com, which already contains additional consumables suggested for the panel.
Parameter values inherited from the design, e.g., number of amplicons or number of pools, cannot be changed. This also includes library kits in bundle with Oncomine Tumor Specific Panels. Only parameters affecting the selection of optional additional consumables are available for editing.
Please refer to the Genexus User Guide for more information regarding how many samples can be run on the Genexus chips.
It is always good practice to run a set of internal performance/validation tests whenever changing experimental conditions for an assay. This includes the use of and/or transition to different sequencing platforms among the ones supported for a specific AmpliSeq panel. Requirements for the testing may vary depending on local regulations or other factors and it is up to the final user of the assay to conduct them accordingly.
Oncomine tumor specific panels were designed to be compatible with degraded DNA such as FFPE, but can also be used on FNA (fine needle aspirate), fresh frozen samples, and other high quality DNA.
Pricing is based on the panel configuration: Manual vs Chef library preparation, reaction pack size and number of genes in the panel.
| Reactions | core | Customized | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Manual | Chef | Manual | Chef | |||||||
| 24 | 96 | 384 | 32 | 128 | 24 | 96 | 384 | 32 | 128 | |
| 1-10 genes | ✓ | ✓ | ✓ | ✓ | ✓ | |||||
| 11-30 genes | ||||||||||
| 31-50 genes | ✓ | ✓ | ✓ | ✓ | ✓ | |||||
| 51-75 genes | ✓ | ✓ | ✓ | ✓ | ✓ | |||||
| 76-100 genes | ✓ | ✓ | ✓ | ✓ | ✓ | |||||
| 101-150 genes | ✓ | ✓ | ✓ | ✓ | ✓ | |||||
The 530 and 540 chips are supported; in addition, estimates are provided for other chips below, based on the number of genes in the panel:
| Chip | GeneStudio | Genexus | |||||
|---|---|---|---|---|---|---|---|
| 510 | 520 | 530 | 540 | 550 | GX5 | ||
| max | 3,000,000 | 5,000,000 | 20,000,000 | 80,000,000 | 130,000,000 | 60,000,000 | |
| min | 2,000,000 | 3,000,000 | 15,000,000 | 60,000,000 | 100,000,000 | 48,000,000 | |
| avg | 2,500,000 | 4,000,000 | 17,500,000 | 70,000,000 | 115,000,000 | 54,000,000 | |
| Core Panels (15-30) | - | - | 8 | 32 | - | - | |
| Oncomine HRR Pathway Predesigned panel (28) | - | - | 6 | 22 | - | - | |
| 1-10 genes | 10 | 2 | 4 | 20 | 81 | 133 | 62 |
| 11-30 genes | 30 | - | 1 | 6 | 27 | 44 | 20 |
| 31-50 genes | 50 | - | - | 4 | 16 | 26 | 12 |
| 51-75 genes | 75 | - | - | 2 | 10 | 17 | 8 |
| 76-100 genes | 100 | - | - | 2 | 8 | 13 | 6 |
| 101-150 genes | 150 | - | - | 1 | 5 | 8 | 4 |
| Chip | GeneStudio | Genexus | |||||
|---|---|---|---|---|---|---|---|
| 510 | 520 | 530 | 540 | 550 | GX5 | ||
| max | 3,000,000 | 5,000,000 | 20,000,000 | 80,000,000 | 130,000,000 | 60,000,000 | |
| min | 2,000,000 | 3,000,000 | 15,000,000 | 60,000,000 | 100,000,000 | 48,000,000 | |
| avg | 2,500,000 | 4,000,000 | 17,500,000 | 70,000,000 | 115,000,000 | 54,000,000 | |
| Core Panels (15-30) | - | - | 4 | 16 | - | - | |
| Oncomine HRR Pathway Predesigned panel (28) | - | - | 3 | 12 | - | - | |
| 1-10 genes | 10 | - | - | 6 | 24 | - | - |
| 11-30 genes | 30 | - | - | 3 | 15 | - | - |
| 31-50 genes | 50 | - | - | 2 | 11 | - | - |
| 51-75 genes | 75 | - | - | 2 | 8 | - | - |
| 76-100 genes | 100 | - | - | 1 | 6 | - | - |
| 101-150 genes | 150 | - | - | 1 | 4 | - | - |
* Assumes 43 amplicons/gene, max number of genes per pricing tier and 2000x depth. For DNA with RNA, 2M reads per RNA library.
Shelf life: 730 days based on the gene with the earliest manufacturing date.
Shipped: Room Temperature.
Storage: -20° C for longest stability. Avoid freeze-thaw cycles.
COA: As this is a custom product, we can only provide a Certificate of Conformance (COC) upon request, as opposed to off-the-shelf products for which a COA is available for download.
The source of annotations is RefGene and the version that we’re using is version v74.
You should expect each order of a panel to be a new lot number. However, there is a possibility that 2 orders provided on the same SO could be the same lot number.
We are investigating the addition of new tumor types and new genes in the future. Please contact support or provide assay feedback on AmpliSeq.com for any additions you’d like to see, including additional genes. Specific design feedback should be provided using the Give Solution Feedback option in the More Actions drop-down menu on the panel design page. To provide general feedback or request help, use the Assay Design Support link in the footer of each page on AmpliSeq.com.
All genes in stock have been tested together to assure they perform well together in any permutation. The core panels and the genes in these panels have been tested to the highest performance compared to other genes in inventory. The core panels also include corresponding analysis workflows in Ion Reporter software. Customized panels do not include default analysis workflows and will require a copy-edit of one of the default workflows. Predesigned panels are similar custom panels and do not include a default analysis workflow in Ion Reporter software, but can be ordered without panel finalization like the core panels.
The minimum number of amplicons is 24 (12 per pool). The maximum number of amplicons is 5000 or 150 genes, whichever is reached first.
The library kits are the same as those commercially available, simply under a different product number. These library kits are only available with the Oncomine tumor specific panels from AmpliSeq.com.
Core panel content is designed to be most relevant for clinical research of the tumor type based on drug labels, guidelines and clinical trials. The panel can be customized if you prefer to add or replace certain genes.
Associated research genes may also be of interest based on our curation of the relevant literature.
Only panels derived from an existing Oncomine tumor specific panels are currently supported for having the RNA content.
We have added a filter category to your “My Designs” section, under the Oncomine Tumor Specific tab, to include designs that exclusively contain DNA and Fusion support.
During the finalization process, the user will be given the option to select whether the design should be finalized as a DNA only design or a DNA and Fusions design. Making the DNA and Fusions design selection will ensure that the Oncomine solid tumor fusion panel is included, and the design categorize in the right section under My Designs.
Each gene is available as is without modification. Please contact support for help designing amplicons covering only your genomic location of interest. An assay feedback tool is available on the designer – please submit any requests for modification to a gene design.
No, this is not an error and is due to the presence of sample ID amplicons in the DNA pools, a design feature in our Oncomine tumor specific panels. Our user interface shows the number of TARGET amplicons and does not include sample ID amplicons in the panel page display or order files. Consequently, you will see a lower amplicon count in the user interface (panel page) compared to the DNA pool tube labels, which are instead configured to report the total number of amplicons present in the tubes. The correct number of target amplicons and of total amplicons is therefore shown both on the AmpliSeq.com user interface and on the DNA pool tube labels, respectively, for your Oncomine tumor specific panels.
Hotspots are regions in the genome where the risk for biologically relevant genetic mutations is elevated (eg. InDels and SNPs). Internally we have curated a list of relevant hotspots that are annotated in Ion Reporter. Amplicons were designed to optimally cover these hotspots. Some genes have amplicons covering the entire gene: other genes have amplicons designed to cover all relevant hotspots, but not necessarily the entire gene. The hotspots are displayed in the new track on AmplISeq.com, with expected amplicon coverage.
We do not currently offer a spike-in function for the Oncomine tumor specific panels. All genes have been designed with respect to each other in order to enable high performance panels, core or customized. We cannot guarantee performance of the panel when a MTO spike-in is used. If your gene is not present, please contact support and we will attempt to include it in the next phase of genes. An assay feedback tool is available on the designer – please submit requests to add new genes to inventory.
Design finalization involves generation of all of the final panel files. Some designs may take longer than 30 minutes. Contact support if the process is not complete within a couple of hours.
All genes, whether CDS or hotspot, have been designed with sufficient amplicons to enable CNV detection. BTK, a hotspot only gene, is the only exception.
The tumor-specific panels are supported on the 530 or 540 chips on the Ion GeneStudio instruments. Manual library preparation is supported with the Ion AmpliSeq Library kit Plus and dual barcodes. Chef library preparation is supported with the Ion AmpliSeq for Chef DL8 kit and IonCode barcodes. Dedicated analysis workflows for the tumor-specific panels are available in Ion Reporter. Generation of Reports is supported in Oncomine Reporter.
We recommend running on the 530 and 540 chips. If you wish to exploring running on a different chip, you will need to create a custom CNB (copy number baseline), SVB (sequence variant baseline) and hotspot file and upload to a custom Ion Reporter analysis workflow. See the Oncomine tumor specific panel User Guide (MAN0018937: Ion Torrent Oncomine tumor specific panels) for the detailed protocol and ask your FAS/FBS rep for support if needed.
You will need to make a custom Ion Reporter analysis workflow using the BED file from AmpliSeq.com as well as create a custom CNV baseline. See the Oncomine tumor specific panel User Guide (MAN0018937: Ion Torrent Oncomine tumor specific panels) for the detailed protocol and ask your FAS/FBS rep for support if needed.
Oncomine tumor specific assay analysis results may indicate a CNV at chr3:193207277 of 116bp in length and not associated with a gene. It is not a CNV hotspot but a sample ID amplicon and should be disregarded.
You can detect exon deletions for BRCA1 in the Oncomine BRCA Expanded panel, or a customized panel thereof, by analyzing your sample with the Oncomine BRCA workflow in Ion Reporter 5.10 or 5.12. You should first run the IR Oncomine BRCA Extended workflow to detect SNPs, Indels, and gene level CNVs in all the genes in your panel. After that, you should run the IR Oncomine BRCA workflow to examine the exon deletions in BRCA1 and BRCA2. We are working to integrate the two workflows into a single workflow in a future version of Ion Reporter, to launch after IR 5.12. Note, CNV baselines can always be augmented with your own samples to increase the number of samples passing QC.
For this version of the software, we’ve set an ordering limit to 500 genes or 15,000 amplicons per panel due to manufacturing restrictions.
Since the order limit is set at 500 genes per panel, it becomes impractical to allow a large number of genes into the Grid or Table view, which will need to be deselected in order to make the design orderable. For this reason, we’ve introduced a limit on the number of genes that can be added to an On-Demand panel.
Yes, an On-Demand design can be edited after it has been created as long as it has not been locked by one of the following actions:
This is different from Made-To-Order designs, which can only be edited in the “Draft” mode and become locked once the job has been submitted and the Results reported.
By finalizing an On-Demand design, the user is agreeing to locking the design from further editing. Once done, user will be able to download panel files (including target bed file), review panel design information, and place an order/request a quote for the panel, but not to edit the design content.
Note: only checked genes will be submitted to the finalized design.
The content of a locked On-Demand panel may be edited creating a panel clone by clicking on the Clone Panel button. A new unlocked panel with the same content, but under a new IAD code, will be automatically created.
Yes, select the “Export targets” button to download the list as a CSV file. This will export all the selected targets displayed in the user interface.
Not directly. Currently, we do not allow the addition of DRA content or hierarchy levels to an existing design, only when the design is being created. The solution is to create a new design with the desired DRA content or hierarchy levels and add content from inventory as desired when in the unlocked design state.
When you click the “Order” button, only genes that are available as On-Demand genes, and which you selected (green), will be ordered. If you create a Spike-in panel, that panel needs to be submitted and ordered separately by visiting the results page of that panel. The Spike-In panel is processed as a made-to-order custom panel and follows the same design submission and ordering process, including separate manufacturing and shipping.
No, once you’ve placed an order, the design cannot be edited because the necessary files needed for analysis by Torrent Suite and Ion Reporter Software need to remain in sync with the material you ordered. If you need to edit your design, select the “Clone” option to copy the panel design. A new IAD number will then be assigned to your design, and you will have the option to edit the design content. Cloning a panel will copy the entire design, selecting any available genes from inventory including any from the Spike-In panel that is now available from potential inventory updates.
Yes, you can always go back to your ordered design and place a new order. If you want to order multiple copies of a panel, the solution is to either order a larger reaction size of the panel (ie one 96 rxn vs three 24rxn) or to go back to your design and add to cart again.
The source of annotations is RefGene and the version that we’re using is version v74.
No, only the coding DNA sequence (CDS) region of a gene is included as part of an On-Demand gene design. If UTRs or amplicons are desired, please contact the support team for potential spike-in solutions via Made-to-Order pipeline.
No, only genes containing CDS regions are supported. At this time, pseudogenes are not supported.
The padding for every On-Demand gene design is either 5 bp or 25 bp on the 5’ and 3’ ends.
Yes, we do support sharing. Alternatively, you can also export the list of targets, and share that list with your collaborator. The design they create will be identical to yours if the list of targets is the same.
The “Score” ranks the relationship between a gene and a disease and it is used to associate genes to disease categories in the Disease Research Areas tool. It takes into account both the strength and number of gene-disease pairs. The algorithm to determine scoring is proprietary. Once genes are added to an AmpliSeq On-Demand panel, they are sorted by the value of this score by default. If needed, switching to Table view on the panel page allows to visualize the Score value for each gene in the panel.
“In-silico” coverage is defined by the percentage of bases that are covered by the tiling of amplicons. This number is a computer-based calculation and should not be confused with experimental coverage, which represents the actual performance of the panel in the lab. We have wet-lab tested all inventoried content in-house.
The number of reads spanning is counted for each base across all padded coding exons of a gene. An average value is calculated for all the bases, and the percentage of bases with read counts above 20% of the average value is defined as “Gene Uniformity”.
No, the number of possible combinations is astronomical and it is not possible to test for all possible combinations in the lab. What our in-house R&D team has done is use computer-based searches to reduce as much as possible the occurrence of primer-primer interactions. The risk is not negligible but deemed very low, backed further by the number of satisfied customers. We have observed << 1% amplicon drop-out due to suspected primer-primer interactions.
Further, we cannot guarantee specifications regarding off-target. We support the in-house GBU and coverage indicated in the IGV viewer and will do our best help troubleshoot if there are any issues that we believe are due to the design or manufacture or our panels.
The default sequencing protocol for On Demand panels is 200bp and 550 nucleotide flows on any chip. If the user wants end to end reads on amplicons greater than 325bp (which can increase detection and accuracy of variant calls by reading both strands), we recommend increasing the number of flows to 650 and using 510, 520 or 530 chips. Amplicons between 275bp and 325bp can use the default workflow. Note, with >550 flows, only one sequencing run per S5 initialization will be possible. The user can determine the size of the insert from the bed file and then identify the amplicons greater than 325bp by adding on the length of the adapters/barcodes (~48bp).
Refer to the Ion AmpliSeq Library Kit Plus User Guide (MAN0017003) for more information.
Shelf life: 730 days based on the gene with the earliest manufacturing date.
Shipped: Room Temperature.
Storage: -20° C for longest stability. Avoid freeze-thaw cycles.
COA: As this is a custom product, we can only provide a Certificate of Conformance (COC) upon request, as opposed to off-the-shelf products for which a COA is available for download.
As these are custom panels, made upon receipt of the order, the turnaround times can vary depending on geographical location but we aim for 2-3 weeks. We have no guarantees for TAT.
We currently do not offer On-Demand panels in larger reactions packs than 32 rxn for Chef or 96 rxn for manual.
In order to obtain proper performance, some amplicons are added multiple times into their respective pool. This is by design. AmpliSeq on-Demand panels that include such amplicons will have this discrepancy. This is not an issue. The panel content is as expected, and the functional number of amplicons is the one displayed on panel page and designed.bed file.
The sources include DisGeNET , Unified Medical Language System and Medical Subject Headings (MeSH).
An in-house gene scoring algorithm was used to create these associations. Details of the algorithm are proprietary but have been described at various national conferences.
No, a preview of the gene content is not available at this time. You need to create the design to view the gene content, but after the design is created it can be reviewed and revised before placing an order.
No, gene content cannot be pre-selected. You can only select full Disease Research Areas categories by clicking on the box on the right, and then edit the gene content once the design is in the On-Demand Grid or Table views.
The number in parentheses ( ) denotes the number of genes in that hierarchical level.
Gene counts often don’t add up as the sum of the subcomponents because one or more genes can belong to multiple Disease Research Areas.
Genes are included in the lists based on their degree of association to a particular Disease Research Area by our algorithms that have aggregated the data. If your favorite gene is not present, it is likely because the observed associations are below our threshold, outside of the sources we used, or did not meet our strict in silico or wet lab testing specifications.
American College of Medical Genetics and Genomics (ACMG) Recommendations for Reporting of Incidental Findings in Clinical Exome and Genome Sequencing.
These are genes associated with conditions listed in the Recommended Uniform Screening Panel (RUSP) for newborns.
Under "Neoplasms", you will see a section for "Neoplasms, hereditary". Here you will find various hereditary oncology malignancies. Note that AmpliSeq On-Demand panels are intended and supported only for inherited disease applications using whole blood (not somatic cancer research with FFPE tissue samples).
We currently only have a search function to find your disease of interest. We do not currently support a back-fill of the hierarchy based on disease selection.
The Integrative Genomics Viewer (IGV) is a visualization tool for interactive exploration of genomic data created by the Broad Institute. Information can be found on their website. The results shown for each gene are from the in-house wet-lab quality control testing (S5, 530 chip). The green track shows regions of the genes where amplicons were designed to target for amplification. The yellow track shows the resulting coverage obtained after sequencing. Users can zoom in to a target of interest and infer expected coverage.
The “Expected coverage” track reflects the number of reads that were observed for each amplicon of each targeted gene during our validation experiments. This track should only be used as general guidance of the likely performance observed when running the experiment. Values are likely to be different when a new assay is performed, but the general coverage trend should remain.
The “Missed regions” are regions where tiling of a high specificity amplicon was not possible due to local environment complexity. We have made every effort to minimize the occurrence of these regions in our On-Demand designs.
The Y-axis represents the experimental coverage, which has been normalized to 100.
No, the IGV viewer has been limited to focus on your gene of interest. In the Grid View, click on a gene and the IGV viewer will be updated automatically and centered on that gene. This version of the IGV viewer is not searchable by coordinate, variant or ID.
All amplicons in the design contain reads that are visualized in the “Expected coverage” track. If reads are not present, they will be highlighted in the “Missed regions (if any)” track. It may happen that, if the number of reads covering an amplicon is relatively small in comparison to neighboring amplicons, the “Expected coverage” track appears empty. However, if you change the scale to a lower value, you will then be able to visualize the lower number of reads.
In order to achieve the most coverage (sensitivity), there is a sacrifice on specificity. So in some instances primers may either bind less tightly or bind off-target, thereby reducing the number of amplicon reads at the desired region.
Spike-in panels are high concentration Made-to-Order panels that are used to expand the panel content to include genes not currently available in On-Demand inventory. Select the “Learn more” link in your design page for more information. Note that we cannot make any guarantees on the performance of Spike-In panels or On-Demand panels when used in conjunction with a Spike-In panel. Although the risk for primer interaction is low, due to the nature of the panels being designed and manufactured separately and combined by the user, we cannot assure performance. Note that Made-to-Order panels are not wet-lab tested by our in-house R&D. We will do our best help troubleshoot if there are any issues that we believe are due to the design or manufacture of our panels.
Since the number of genes available as On-Demand genes is limited, a Spike-in panel enables a user to sequence all the targets initially wanted in a single target amplification reaction. Note that a Spike-In panel is limited to < 123 amplicons per pool, or 246 amplicons total for a 2 pool On-Demand panel.
The limitations of Spike-in panels involve the number of genes that can be included and the loss of the performance guarantee. The size of a compatible Spike-in panel is limited to 123 amplicons per pool, for a total of 246 amplicons. Any designs exceeding this limit cannot be designed from the On-Demand panel design page and are not supported as the On-Demand panel performance may suffer due to dilution effects. From a performance standpoint, since Spike-in panels are manufactured as Made-to-Order panels and are not wet-lab tested like On-Demand panels, we cannot guarantee performance. Adding a Spike-in panel to an On-Demand panel will void the guarantee and should be done only if the user accepts this limitation. We suggest the user spike-in to a smaller number of On-Demand reactions and test on known samples before continuing to a larger number of samples.
Spike-in panels follow our Made-to-Order process. They are synthesized de novo at every order and are not wet-lab tested nor do they have any performance guarantee. Depending on the number of amplicons, Spike-In panels are offered in 750 reactions (<= 96 amplicons) or 3000 reactions (> 96 amplicons).
On the other hand, On-Demand Panels have optimized designs, have been pre-manufactured, and wet-lab tested. They are available in small reaction number batches (8 or 32 for Chef and 24 or 96 for manual). On-Demand Panels also contain data that can be visualized in our IGV viewer available on the design page.
PKD1
PKD1 is a challenging gene to sequence due to allelic heterogeneity, high GC content, and homology of the PKD1 gene with six pseudogenes resulting in inability to discriminate between homologous regions of PKD1 and its pseudogenes. This gene may increase off target reads. Variants found with this design that have low mapping quality should have concordance testing performed to determine exact location of the variant.
MYH6 with MYH7
The designs for MYH6 and MYH7 will perform as expected when alone; however, in a panel with both genes present you may see an increased frequency in unusually long and short amplicons due to inter-gene priming resulting in poor end to end reads in certain exons of the genes. If this is highly undesirable, our White Glove team can assist in optimizing a design for a panel containing both these genes.
CFTR or DMD
AmpliSeq On-Demand designs cover the CDS region of a gene. Some mutations, such as genomic rearrangements, large deletions and intragenic rearrangements and intronic variants may not be identified by this assay since enrichment is focused on the exons, and use of a complementary assay is recommended.
PMS1 or PMS2
PMS2 is a homolog of PMS1. Since these genes have high homology, unique mapping is very challenging. We design genes so that the amplicons have at least 2 mismatches to the next best alignment, however in some cases low mapping quality alignment may be observed. In such regions, we recommend Sanger sequencing or long-range PCR to confirm variants.
It depends on the application. If you have an application that requires ultra-high sensitivity, then ASHD is recommended. If you have applications that require high multiplexing, then regular AS is the preferred option.
ASHD currently supports up to 4999 amplicons (9999 oligos) per design. This limit is enforced in AmpliSeq.com.
The approximate number of reactions worth of material provided for ASHD panels is 3,000 reactions for 1-pool panels and 6,000 reactions for 2-pool panels (manual library preparation).
Germline applications may be considered in the future, depending on the relevance use of ASHD technology.
The currently supported variant types are, SNVs, small Indels, Fusions from a pre-designed list, and CNV through design best practices available in AmpliSeq.com.
In this case, a request to the AmpliSeq Custom Services team will need to be submitted. Contact your local sales or support representative to learn more.
Yes, the AmpliSeq Custom Services team will be supporting specialty designs for ASHD. Submission of specialty ASHD designs to the AmpliSeq Custom Services team should follow the same path for AS design requests, which involves contacting your sales or field support contacts for help.
Automated gene expression designs are not currently supported in AmpliSeq.com. However, custom gene expression designs can be created by the AmpliSeq Custom Services team. Contact your local sales or support representative to learn more.
Yes, but only through our AmpliSeq Custom Services team. Designs created in AmpliSeq.com are limited to 1-pool for DNA hotspots, 1-pool for RNA fusions, and 2-pools for DNA gene designs.
The primers used in ASHD are more complex than for regular AS, so it has been recommended to keep them in separate pools for storage and long term stability. FWD and REV primers should only be mixed at the time the libraries are created. Please refer to ASHD Library Kit User Guide for more information.
Yes, the chip selection is governed by the following two considerations:
Chip throughput requirement to achieve the desired LOD and the number of samples per chip for the specific assay panel with the specific number of amplicons. Refer to Chapter 6 of the User Guide (MAN0017392) to learn more about the approximate number of libraries supported per chip type based on the number of amplicons per library. See Appendix C to learn about the coverage read requirements based on input amount and target LOD.
Lengths of amplicon for AmpliSeq HD designs supported by different chips. For cfDNA/FFPE designs with amplicon size at 75-125bp, your assay is supported on the 530, 540 and 550 chips. However, for the FFPE only design with amplicon size at 125-175bp, your assay is officially supported by only the 530 chip. If higher throughput is needed, 540 can be used but may yield lower number of aligned reads (~50 million reads) on a 540 chip. We do not recommend the use of the 550 chip for the FFPE only design.
Our studies have shown that as the chip capacity increases, the end-to-end read percentage decreases. Since AmpliSeq HD requires that both barcodes (5’ and the 3’ end) be read correctly, reads that do not have good end-to-end get removed, and hence the total number of families gets lowered and we’re unable to achieve lower limits of detection.
Dual use designs are designs that can be used for multiple DNA types such as DNA extracted from cell free or FFPE stored tissue. In the case of cfDNA/FFPE, small sized designs (75-125bp) have been shown to be suitable for both types of DNA.
Yes, 520 chips may be used, but they required custom Torrent Suite parameters. Default parameters are only provided for the 530 chip; consult your local Field Bioinformatics Scientists for help with setting up the parameters in Torrent Suite for the 520 chip.
AmpliSeq HD panel with greater than 3 pools can be occasionally created in copy amplicon designs (Subset design), in order to resolve primer conflicts. However these solutions cannot be manufactured, hence the Preview order button is disabled. For support on possible primer conflict resolution strategies, please contact support Americas, EMEA, Greater China, South Asia, Japan for assistance.
Refer to the link for a table describing the number of reactions that you can expect, depending on the size of your panel.
Refer to the link for a table describing the number of reactions that you can expect, depending on the size of your panel.
Refer to the link for a table describing the number of reactions that you can expect, depending on the size of your panel.
No, not currently. The only format available for RNA designs is in Tubes Only format.
To maximize convenience and flexibility. Pooled primers can be used immediately. Plated oligos can be used to: 1) Rebuild the same pool, 2) Rebuild a pool with fewer primers.
Currently, the only way to do this is to duplicate the target list in a new version, add the new genes and resubmit. As long as the same design attributes are set (CDS/all exons 150/200) as previously used, the genes from the previous set will have the same design.
This depends. With the exception of orders from Europe that are processed in the U.K., all other international orders are first processed by the North America customer service team, who sends the form to the local customer service team for verification and final changes. The local customer service team works with the customer to determine the best route for a shipment, and the decision is made by the local customer service team. Shipment to a distribution center is slower, but it is significantly less expensive for Thermo Fisher, and could potentially result in fewer customs issues and tax charges. Direct shipment is generally faster, but this adds additional shipping costs for Thermo Fisher, and there may be customs and/or tax implications.
Yes, you will need to use the "Copy Amplicons" function to ensure the amplicons used in the original design are used for your new design.
Yes, you can add amplicons to your assay using the process known as "Spike-In". Check with your local Field Support team for guidance on how to add the new targets, and make sure you make the selection for 50X primer concentration at the time of ordering.
Ion AmpliSeq Custom Panels range from 12 amplicons* to 6,144 amplicons per tube. The minimum of 12 amplicons is due to chemistry performance limitations with low number of amplicons per pool. The exception to this limitation are spike-in designs which allows < 12 amplicons/pool, since these panels are exclusively meant to be used to physically add amplicons to larger main designs to expand their content. Note: if an MTO DNA design panel has fewer than 48 amplicons it is subjected to our minimum order quantity policy of 48 amplicons (96 oligos) and its associated pricing.
*Target regions can be as small as 1 kb and can go up to 5 Mb.
Both. Each custom primer pool is delivered as both a pre-pooled tube and as individual primer pairs plated into 384-well plates. Small orders of up to 96 amplicons per pool will contain 750 pre-pooled reactions and individual primer pairs sufficient for 1,500 reactions. Larger orders of more than 96 amplicons per pool will contain 3,000 pre-pooled reactions and individual primer pairs sufficient for 6,000 reactions.
Email us at genomicorders@thermofisher.com or call 1-800-955-6288, x46636. Please use your Ion AmpliSeq Design ID number when referring to your order. Please contact your local customer service outside of North America.
There is no guarantee of cancellation of a custom oligo order. Please contact your local customer service representative for more information and options. On occasion, customer service can intercept an order and is able to cancel it prior to synthesis. You must call ASAP: 1-800-955-6288, x46636.
Refer to the link for a table describing the number of reactions that you can expect, depending on the size of your panel.
Refer to the link for a table describing the number of reactions that you can expect, depending on the size of your panel.
The only order options available are, either Tubes Only or Plates Only option. If you need to order Tubes and Plates, we recommend placing 2 separate orders.
The ordering process and the format/content of AmpliSeq panels remain unchanged. The Order Preview page shown at the end of the preview order process includes both technical and ordering details meant to provide a detailed summary of the specific selections made before proceeding with the actual ordering of the panel (optionally, a list of additional recommended consumables based on the selected panel configuration can also be displayed and desired items can be added to the order as needed). Once you are satisfied with the panel format configuration shown in the Order Preview page, depending on your account setup on thermofisher.com and your local/administrative regulations, you can either order your AmpliSeq panel directly online through the Thermo Fisher Cart (“Add to cart” button) or place an offline order (e.g., through a written purchase order) through your usual Customer Service channels based on the information provided on the same page. * The Download SOS button at the bottom of the Order Preview page allows capturing and exporting in a text file (SOS Document) all the relevant information related to the chosen panel configuration. The SOS document is required when placing an order that is processed by Customer Service teams (e.g., purchase order sent over email). In general, when attached to offline communications with Thermo Fisher Scientific personnel, the SOS Document can be used to facilitate the correct processing of your requests for the specific panel configuration of your interest (e.g., requesting quotes, raising technical support inquiries, etc.) Orders placed in the Thermo Fisher Cart, do not require the SOS document.
Customer Service inquiries related to AmpliSeq orders can be sent by email to the following addresses (by region):
Americas: genomicorders@thermofisher.com
EMEA: uk.primers@thermofisher.com
Asia Pacific: orders.sg@thermofisher.com
Japan: jpprimerorder@invitrogen.com
China: ampliseq_cn@lifetech.com
*Different processes might be in place in countries served by distributors, which are also unchanged. Please refer to your usual local Thermo Fisher Scientific resources for more information.
There has been no change in format, manufacturing process or materials delivered for any AmpliSeq panel, including Made-To-Order, Community and HD. The “Provided materials” field in the Order Preview page is meant to provide specific details on the delivered materials for the panel format chosen by the user (in addition to the general information for standard Made-To-Order and HD formats reported in this same FAQ section). Among other things, this information, combined with the AmpliSeq reaction setup details available in the pertinent User Guide (direct link also provided in the Order Preview page based on the panel format selection), is meant to be used to estimate the number of samples/reactions that can be processed given the selected panel format and library preparation method. Please contact your Thermo Fisher Technical Support resource in case help is needed in this respect.
The information in the “Chosen Format” field is a text string meant to provide confirmation of the selection made by the user on the “format” button and on the “special instruction” menu during the order preview journey. For Made-To-Order and Community panels, the reported info is “format button, special instructions”; for AmpliSeq HD panels, only the “format button” info is reported (no special instructions menu available).
Note: For Made-To-Order panels, the “384-well plates only” special instructions menu entry is available under the “Tubes plus 384 well plates” format category button. Therefore, according to the above, the “Chosen Format” field for this selection is “Tubes plus 384 well plates, 384-well plates only”. As per the corresponding “Provided materials” field, only 384-well plates will be provided for this selection.
The Design ID defines the content of an Ion AmpliSeq™ custom panel. For certain panel types – including Ion AmpliSeq™ Pre-Designed Panels, Ion AmpliSeq™ White Glove panels, and Oncomine™ tumor-specific Core panels – the same Design ID may be ordered by multiple users. In these cases, an additional alphanumeric suffix (preceded by “!”) may be appended during quote generation or order processing to create a unique transaction reference.
This suffix does not change the panel content or the selected order configuration. Panels that share the same Design ID correspond to the same underlying design, regardless of any appended suffix.
To submit human or mouse genomic targets for assay design submission, users can input a BED file of genomic regions of interest, or a Gene List file based on HUGO gene symbols and aliases.
The BED format files in AmpliSeq use the convention known as “zero-based, half-open” (ZBHO) coordinates, both for input and for output files. In contrast dbSNP and COSMIC use “one-based, inclusive” (OBI) coordinates. Notice then that compared to dbSNP and COSMIC, AmpliSeq coordinates will have a start coordinate one less than that shown on the dbSNP and COSMIC databases.
When comparing coordinates in BED files between AmpliSeq and data from the UCSC browser, please be aware that the UCSC Genome Browser uses both coordinate systems: OBI in the web interface and ZBHO in their database and data downloads.
A design of 250kb or less should be returned in less than 48 hours of submission. For designs over 250kb or a large number of targets, you should expect a longer turn around time.
Yes, you can generate BED formatted files by utilizing the UCSC Genome Browser export feature in the Table Browser section.
Not at this time. We are currently exploring different methods for uploading regions to the Ion AmpliSeq Designer.
Yes. Any tools can be used to help you generate files for submission, but it is important to make sure the correct version of the genome is being used (hg19 for human, mm10 for mouse).
We support Firefox, Google Chrome, Safari, and Internet Explorer 9 and above.
If coverage obtained from the initial design is less than 100%, you can try to extend the primer further out into the intron to capture the whole exon. Primer regions are not considered covered, so placing padding may ensure that we are able to get good quality sequence at the ends of exons, and to get some sequence read into the splice junction regions.
Yes, Ion AmpliSeq Designer allows you to do SNP genotyping by sequencing. Alternatively, you can also consider Taqman SNP Genotyping Assays for a large number of samples.
The Ion AmpliSeq Designer does not currently allow designing customized primers for methylation experiments. However, our White Glove team can assist creating a custom methylation panel for that purpose. Please contact your local representative for more info.
The Ion AmpliSeq is used for targeted resequencing. It cannot be used to sequence whole genomes.
When you click on the Download Results button of your Results ready project/version, the following output files are generated in a compressed folder.
| File Name | Details |
|---|---|
| #_coverage_summary.csv | Gene-specific and region-specific coverage details |
| #_coverage_details.csv | This file provides details of coverage by exon for targets submitted by CDS or CDS+UTR (targets submitted as regions cannot be decomposed into exon-equivalents, so they are not listed in this file). If a request has no CDS (or CDS+UTR) targets, then there is no information for creating this coverage_details.csv file. |
| #_Submitted.bed | BED file with the genomic coordinates submitted to design |
| #_Designed.bed | BED file of coordinates of what the application designed to |
| #_Missed.bed | BED file of coordinates that were missed by the designer |
| #_384WellPlateDataSheet.csv | This file provides the position of amplicons (or single primers) in the 384-well plates that are included with tubes and plates panel order formats (compatible panels only). For each amplicon or primer in the panel, the file reports the row and column coordinate of the corresponding well in the plate, along with info on reference, chromosome, amplicon insert start, and amplicon insert end. Empty plate wells have the value "Blank" in the "Amplicon_ID" column (and the other fields are left empty). |
| #_hotspot.bed | This file contains all hotspot variants that overlap with designed.bed coordinates (specific to each design) and an internally curated list of oncology hotspots. This file is available by default for AmpliSeq HD designs (IAH) and Oncomine Tumor Specific Panels (OAD if customized), and can be optionally generated for human custom DNA Made-To-Order designs. |
| #_amplicon_insert_size_histogram.png | This image file provides the user with a histogram view of the insert-size distribution of all the amplicons found in the panel. The insert being the region between the primers, which is amplified as reads, which in turn are used for sequencing of the targeted region. |
| #_amplicon_size_histogram.png | This image file provides the user with a histogram view of the amplicon size distribution of all the amplicons found in the panel. The amplicon being the targeted region including the primer information at the 5' end and 3' end. |
| plan.json | This file contains information to automatically configure a run plan for the panel, when the panel's files are directly downloaded from the Torrent Server 3.6 |
AmpliSeq primers manufactured by Thermo Fisher are optimized for their use with the Ion Torrent platforms. The primer sequences represent confidential information that cannot be disclosed to users or to any third party under any circumstance.
The user interface does not check for duplicate regions or any overlaps of the regions submitted in a .bed file. The UCSC .bed files typically contain duplicate regions for many quasi-identical transcripts. Too many overlapping regions may lead to a wrong estimate which may prevent the submission if the target size exceeds the currently allowed limit of 500 Kb.
A simple and effective way that may help to prevent this, is by running the UCSC .bed file through the program mergeBed from the BEDTools suite. This will create equivalent regions in a smaller .bed file.
The largest design that can be submitted directly to the pipeline is at most 500 Kb. However the pipeline is capable of processing designs up to 5 Mb, but such designs are predictably costly and take up a large number of computational resources.
In the cases of submissions larger than 500 Kb, the user will be contacted by email requiring more details about his/her interest in that particular, design and the design will be put on hold until the contact has been made.
The DNA made-to-order pipeline in AmpliSeq 7.2 incorporates a number of algorithm improvements developed within the Custom Solutions Group at Thermo Fisher. These improvements have been validated on a large number of designs, and provide improved coverage and accuracy.
In the majority of cases, AmpliSeq.com will provide more coverage than previous versions. However, the new algorithm also implements additional checks and the quality of candidate oligos and amplicons, in particular, checking for specificity and insert uniqueness. In some less common scenarios, for example in cases of low-complexity regions of DNA, the algorithm may not be able to find amplicons meeting these quality checks to cover a region. In this situation, the reported coverage may be lower than previous versions, though actual amplicon performance should be improved.
AmpliSeq.com 7.2 will continue to generate multiple solutions corresponding to different stringency levels and numbers of pools. However, we have found that generating solutions with different target amplicon lengths were not needed in most cases. Consequently, AmpliSeq.com will now only generate solutions for the optimal amplicon length for the desired sample type. This only applies to AmpliSeq DNA made-to-order. See the next question for further details.
In 7.2 the changes will affect AmpliSeq DNA made-to-order releases for human (hg19, GRChg38) and mouse (mm10) genomes. Improvements for additional genome support, including support for custom genomes, and support for AmpliSeq HD, are planned for a future release.
No. Once you're ready to submit your design for calculation, you will be prompted to select the desired amplicon size, and only solutions for the selected amplicon size will be generated.
When using the copy amplicons functionality, the system will attempt to create a solution which includes all the requested amplicons, if necessary adding additional pools, up to a maximum of 5 pools, in order to accommodate overlapping amplicons. If it is not possible to create a solution using the maximum number of pools then an error is raised. In this case the user must remove some overlapping amplicons/targets in order to create a valid solution.
Region names can be at most 70 characters long, and may not contain any of the characters '/', '"', ''', '=', ',', ';', '[', ']', '\', '|', space or tab.
Uploaded target bed files must have the extension '.bed' (lower case). There are no other restrictions.
If a gene-symbol is not currently in the known list of aliases then it will be necessary to upload the specific regions included in the gene, in order to design a solution for the gene.
Yes, please refer to the following compatibility table to understand the changes done during the Copy amplicons event:
| Source DNA Type | Initial Draft DNA Type | New Modified Draft DNA Type |
|---|---|---|
| cfDNA (140bp) | cfDNA (140bp) | No modification |
| cfDNA (140bp) | FFPE (175bp) or Standard (275 or 375bp) | No modification |
| FFPE (175bp) | cfDNA (140bp) | FFPE (175bp) |
| FFPE (175bp) | FFPE (175bp) or Standard (275 or 375bp) | No modification |
| Standard (275bp) | Standard (275bp or 375bp) | No modification |
| Standard (275bp) | cfDNA (140bp) or FFPE (175bp) | Standard (275bp) |
| Standard (375bp) | Standard (375bp) | No modification |
| Standard (375bp) | cfDNA (140bp) or FFPE (175bp) or Standard (275bp) | Standard (375bp) |
Yes, please refer to the following compatibility table to understand the changes done during the Copy amplicons event for AmpliSeq HD:
| Source DNA Type | Initial Draft DNA Type | New Modified Draft DNA Type |
|---|---|---|
| cfDNA (125bp) | cfDNA (125bp) | No modification |
| cfDNA (125bp) | FFPE (175bp) | No modification |
| FFPE (175bp) | cfDNA (125bp) | FFPE (175bp) |
| FFPE (175bp) | FFPE (175bp) | No modification |
Yes, please refer to the following compatibility table to understand the changes done during the Copy amplicons event where the draft design is for AmpliSeq HD:
| Source DNA Type | Initial AmpliSeq HD Draft DNA Type | New Modified Draft DNA Type |
|---|---|---|
| cfDNA (140bp) | cfDNA (125bp) | FFPE (175bp) |
| cfDNA (140bp) | FFPE (175bp) | No modification |
| FFPE (175bp) | cfDNA (125bp) | FFPE (175bp) |
| FFPE (175bp) | FFPE (175bp) | No modification |
| Standard (275 or 375bp) | cfDNA (125bp) | Only amplicons <200bp will be copied, others will be ignored. Draft will be converted to FFPE type (175bp) to accommodate as many amplicons as possible. |
| Standard (275 or 375bp) | FFPE (175bp) | Only amplicons <200bp will be copied, others will be ignored. Draft will remain as FFPE type (175bp). |
No, it is not possible to copy amplicons from an AmpliSeq HD design to a regular AmpliSeq design.
Coordinates in the Polymorphism.bed file indicate regions of the sequences in the custom reference FASTA file with high polymorphism (i.e., SNPs, indels, or other variation). Ion AmpliSeq Designer avoids overlapping primers with such regions since performance may be unpredictable. Consequently, in regions with many polymorphisms, Ion AmpliSeq Designer may fail to find suitable primer locations in order to sequence these regions. In this situation it may be necessary to resubmit the Polymorphism.bed file with very rare polymorphisms (e.g. minor allele frequency < 1%) removed.
The Copy Amplicons function ensures that all amplicons copied from a source design are included in the new design. The assignment of amplicons to pools is re-generated for each design with the aim of balancing the pools and avoiding any inter-amplicon conflicts. Balancing of the pools ensures that you have a similar number of amplicons in each pool. As a result, whenever amplicons are added to, or removed from a design, the pool assignments of other amplicons within the design is likely to change.
No. A hotspot BED file defines region targets that typically represent relevant variants. Specifying a hotspots file to use in a run is optional and enables Torrent Variant Caller to identify and report if a specific variant is either present, absent or could not be determined in the positions defined in the file. Specifically, a hotspot file instructs the Torrent Variant Caller to always include these variants in its output files, including evidence for called variants and filtering threshold(s) that disqualified a variant candidate in case a call could not be made. A hotspot file affects only the variantCaller plugin, not other parts of the analysis pipeline. If you don't specify a hotspots file, the software output includes only the variants detected between your sequence and the reference genome.
Copied amplicons are considered must-have content for a subset design, therefore they will always be included in the final design. In case of conflict with additional newly designed amplicons (including amplicons for CNV gene targets), the copied amplicons will always prevail (i.e., they will be those retained in the final design at the expense of the new amplicons). This could potentially impair the automatic CNV design pipeline in unpredictable ways. Therefore, when a Made-To-Order design is submitted, if copied amplicons are detected alongside with CNV gene targets, then the automatic CNV design functionality is switched off and the warning banner is presented to the user for confirmation of the same.
For the same reason, combining CNV-compatible gene targets with existing amplicons is currently not supported.
It’s not possible to detect CNVs on sex chromosomes if all the targets on a panel are on the sex chromosomes (additional amplicons must also be added to autosomal chromosomes by adding targets accordingly). The automatic gene level CNV design functionality can be used to add CNV gene target on sex chromosome, but it does not automatically check for the presence of additional autosomal targets. Therefore panel-level CNV compatibility when sex chromosome CNV targets are present must be manually verified by the user.
When the user submits a gene to design, only exons are used as targets. If you wish to design across the whole gene (exons and introns) the user needs to submit the start and end coordinates of the gene.
No. The designer uses exon coordinates as listed by the UCSC Genome Browser. Promoters are not part of the exons and need to be requested using a BED file describing the genome coordinates.
Primers in the same tube do not overlap. As our product line evolves this might change in the future and a small overlap might be possible.
The process is an automated pipeline, optimized to provide the maximum coverage with reliable primer sets.
Each primer pool goes through a rigorous process to meet strict design specifications. During the design of our pipeline, we validated a substantial number of our custom assays though wet lab testing.
If an overlapping region is submitted to the design pipeline, internally the region is concatenated and treated as a single region for design, thus there will be no overlap. The two regions are reported back in the UI as submitted. While it is possible that an amplicon might be prorated twice, once in each of the original regions, this amplicon (and its primers) only occurs once in the design (see the plate file).
A superamplicon is created when two forward PCRs joined to form one large amplicon. The pooler algorithm in the pipeline separates primers into separate pools to minimize this.
chromStart - The starting position of the feature in the chromosome or scaffold. The first base in a chromosome is numbered 0.
chromEnd - The ending position of the feature in the chromosome or scaffold. The chromEnd base is not included in the display of the feature. For example, the first 100 bases of a chromosome are defined as chromStart=0, chromEnd=100, and span the bases numbered 0-99.
Development work from over a decade allows us to produce primer designs that allow simultaneous amplification of many amplicon targets. A unique chemistry has been developed for Ion AmpliSeq that allows removal of any primer dimer formed along with the majority of the primer itself from the amplified template. This makes sequencing very efficient by not wasting bases on non-informative primer sequence and allows for very clean sequencing reactions.
Yes. The pipeline first attempts to design primers that only match the target, and not the pseudogene (or duplicate) version(s). If the target gene is not covered in the initial rounds of primer selection, then the match parameters are relaxed, for the sake of coverage, in later rounds, attempting to maintain the uniqueness of the inserts.
The pooling step in the design is optimized in order to minimize the interference between overlapping amplicons. Hence, overlapping amplicons would be segregated into different pools.
There are several reasons that explain why this happens:
DNA
Gene targets correspond to RefGene v98 for Human (hg19) and v99 for Human (hg38)
| Genome | Genome version | dbSNP | COSMIC |
|---|---|---|---|
| Human (hg19) | Feb. 2009 (GRCh37) | v156 | v97 |
| Human (hg38) | Dec. 2013 (GRCh38.p2) | v156 | v97 |
| Mouse (mm10) | Dec. 2011 (GRCm38) | v150 | N/A |
RNA
Human RNA Canonical RefSeq Transcripts* - Feb. 2009 (hg19, GRCh37)
HGNC Database, HUGO Gene Nomenclature Committee (HGNC), EMBL Outstation - Hinxton, European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridgeshire, CB10 1SD, UK http://www.genenames.org, 11/2012
*For compatibility reasons, Spike-in designs use the RefGene version of their corresponding pre-designed AmpliSeq On-Demand panel (v74).
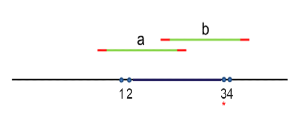
When non-overlapping amplicons covering adjacent submitted hotspot targets are not available for a one-pool design (example figure below), the hotspot pipeline will compare overlapping amplicons ("a" and "b", in the example) and eliminate one or more of them, resulting in the missed coverage of the targets they covered.
The target prioritization functionality allows the user to define a priori which hotspot target(s) the design pipeline should attempt to retain when such conflicts by overlapping amplicons occur. If hotspot targets are prioritized by the user, then the design pipeline will attempt to resolve conflicts by preferentially retaining amplicons, if available, that cover the prioritized targets, at the expense of non-prioritized ones. Although under certain circumstances target prioritization might also resolve conflicts in 2-pool designs, this functionality is mostly relevant for 1-pool hotspot designs where overlapping amplicons are mutually exclusive (i.e., cannot be all retained in the final 1-pool design).
Note: The hotspot target prioritization functionality is not meant to be used for increasing the overall coverage of 1-pool hotspot designs (in case, lower specificity solutions can be used for the scope, see FAQ section), but instead, if suitable amplicons are available for the scope, to try and prioritize the coverage of hotspot targets most relevant for the user when overlapping amplicon conflicts occur due to target proximity.
|
Effect of prioritization: With reference to the above figure, if target 3* is prioritized by the user at the hotspot design submission step, then the design pipeline will retain amplicon "b" for the 1-pool design solution and the prioritized hotspot target 3 will be covered (along with target 4). Amplicon "a" will instead be eliminated resulting in the missed coverage of hotspot targets 1 and 2. |
In general, they are same. The CNV algorithms rely on having sufficient numbers and diversity of amplicon targets with normal diploidy in the sample. However, we do not have a specific minimal number of amplicons-per-panel defined/tested for AmpliSeq Custom or Ampliseq HD Custom CNV calling. General consideration is that for panels with few number of amplicons (less than 10), it is difficult to create/apply a baseline and detect CNV.
Please contact your our Global Support team (ampliseq-designs@thermofisher.com) to request the transfer. Note that the transfer will be done not just for the requested design, but also for all designs associated with the same Custom Reference. This is an all or nothing event.
When transferring a design created from a Custom Reference, all designs must be transferred along with the reference. The other designs should now be found in the recipient account.
a) You can share designs whether they are from Reference Genomes like (hg19, mm10 and others), as well as designs created from Custom References using the “Sharing” functionality found on the main design page, next to the Download results button.
b) After selecting the “Sharing” button, you will need to enter the full email address of the recipient you wish to share the design to. Please note that only registered emails will be recognized as valid recipient email address. Make sure that the recipient does have a valid AmpliSeq.com account.
a) Share designs are meant to be used for review, analysis and ordering purposes. You may not create new designs from a shared design. For this reason, the “Copy Amplicons” and “Copy Targets” functionality is disabled.
b) The sharing of a design can only be done by the original creator of the design. A recipient of a shared design, cannot re-share the same design to another user.
We have decided to adopt NCBI formatted references as our standard for uploading Custom References to AmpliSeq.com. This means that users will be able to download references files from the NCBI databases.
Please visit the help section for Custom References Working with Custom References for further information about all the requirements needed for successfully upload your Custom Reference.
Yes, in our latest release we now allow the removal and re-upload of the Polymorphism (BED) file, without the need for a new Custom Reference.
No, only the latest Polymorphism (BED) file associated with the Custom Reference is available from the Design References page. If the file is removed, it can no longer be accessed or retrieved.
The design sharing functionality has been enhanced to provide our users with better oversight and control of which designs have been shared and who they’ve been shared with.
No, the previously shared design links will no longer work. For collaborators to have access to previously shared designs, the collaborator will need to request access from the creator of the design. The design author will need to re-share their previously shared design using the new sharing functionality.
In the user dashboard, new icons and filtering options have been added to help users manage their shared designs. These icons provide with a view of the collaborators without having to open the design view.
If you need to share the same design with multiple collaborators, you will need to add the user email id separated by comma.
In order to stop sharing a design, you will need to visit the “Sharing” functionality and click the waste bin icon. This action will remove the selected user from having access to the design.
Whether it is done intentionally or unintentionally, when you delete a design your collaborators will no longer have access to the design. Contact our support team if you have unintentionally deleted a design.
At this time only email addresses that have been registered in www.ampliseq.com are recognized as email valid emails. If you would like to share a design to your collaborator, encourage them to visit www.ampliseq.com and register. Before the design has been shared, they will need to log in to the site and accept our Terms and Conditions.
Yes, they are the same version of the human genome. GRCh38 stands for “Genome Reference Consortium Human Reference 38” and it is the primary genome assembly in GenBank; hg38 is the ID used for GRCh38 in the context of the UCSC Genome Browser.
The hg19 build is a single representation of multiple genomes. The hg38 build provides alternate sequences (“alt_sequences”) for some genomic regions for which their variability prevents adequate representation by one single reference.
• The version used by our software is based on GRCh38.p2 (http://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.28)
• Unplaced, Alternate and Unlocalized contigs are listed as separate chromosomes and ordered first by chromosome localization, then by the alphabetic order of the Genbank accession of the contigs.
• Repeat and SNP locations are soft-masked into lower case letters, while the ambiguous IUPAC bases, duplicated centromeric arrays, and chrY PAR regions are hard masked into 'N's.
• It contains chr1-22, chrX, chrY, and chr22_KI270879v1_alt.
• Contig chr22_KI270879v1_alt is hard masked except region 269814-279356 (1-based).
• Gene GSTT1 is located at chr22_KI270879v1_alt:270308-278486.
Our version of hg38 only considers the chr22_KI270879v1_alt. This alt chromosome contains gene GSTT1 that was part of chr22 in hg19. This was an internal decision which was made to enable standardization of the genome reference for use across multiple businesses within our organization.
We strongly recommend that you download our version of the hg38 from our website. This version is the one that is assumed in all of our software applications and has been tested for compatibility.
At this moment we do not offer any conversion tools. We recommend our users touch base with their own bioinformatics experts for further guidance.
No. The old assays and panels were created using hg19 as a reference and should be analyzed with the tools and analysis pipelines created for hg19.
Yes. The pipelines and tools for using hg19 as reference for design and analysis are still available.
Yes. The variant calling workflow based on hg19 will be available in Ion Reporter. If your design was created using hg38, then you can also call and annotate variants using Ion Reporter.
No. Amplicons from a custom design can only be copied to another custom design associated with the same reference. It is not possible to copy amplicons to a custom design associated with a different reference even if both references are human.
Not at this moment. The off-the-shelf panels (ie exome), On-Demand panels and Community panels will still be based on hg19. Conversion of pre-designed panels may be considered in the future based on market demand.
Not at this moment. The Oncomine panels will still be based on hg19.
If your AmpliSeq design has been created using hg38 as a reference, then you can create an ad-hoc workflow in Ion Reporter for analysis. All analysis and annotations will take in consideration hg38 as a reference. However, at this moment there are no hg38 workflows in Ion Reporter. The tools for analysis and the annotations for hg19 will still be available.
The pipeline recognizes HGNC approved gene symbols, previous gene symbols, and synonyms. It is case sensitive. Please check the Gene Symbol entered for typos and then search for the Gene Symbol on the HGNC website , to confirm that is a valid entry. The pipeline requires that the Gene Symbol entered is unambiguous, meaning that it resolves to a single HGNC approved gene symbol.
The pipeline uses strict criteria in order to design a single assay per gene that will result in consistent amplification while minimizing the risk of amplifying genomic DNA, this is done by targeting splice sites between exons. If there is not a splice site that is shared between all RefSeq transcripts of the entered Gene Symbol or if a passing assay could not be designed to the most common splice site then an assay will be produced that is compatible with a subset of the RefSeq transcripts for the entered Gene Symbol.
The pipeline attempts to resolve a single hg19 genomic alignment, where an exception is made for pseudoautosomal alignments, for each RefSeq accession. Unrecognized or Invalid RefSeq transcript accessions can result when the entered accession has been permanently suppressed or was not successfully resolved to a single hg19 genomic alignment.
The pipeline will design a single most-inclusive assay for the gene that corresponds to the entered RefSeq transcript accession. To achieve this, the pipeline will attempt to design an assay to the most common splice site found in the RefSeq accessions for the gene.
No. These are among the list of features that are on the development roadmap and will be included in subsequent releases of the Ion AmpliSeq RNA pipeline.
No. The RNA pipeline does not allow specifying genomic regions for design.
No. The RNA pipeline does not allow the entry of FASTA sequences for design.
The data on which AmpliSeq RNA pipeline is based on, is the NCBI's RefSeq sequences. A consequence of this is that all coordinates in the .BED files correspond to RefSeq coordinates that are not recognized by the UCSC genome browser. However the .BED files produced by the RNA pipeline are fully compatible with other pieces of IonTorrent software.
The minimum of 12 amplicons is due to chemistry performance limitations with low number of amplicons per pool. The exception to this limitation are spike-in designs which allows < 12 amplicons/pool, since these panels are exclusively meant to be used to physically add amplicons to larger main designs to expand their content.
Note: if an MTO DNA design panel has fewer than 48 amplicons it is subjected to our minimum order quantity policy of 48 amplicons (96 oligos) and its associated pricing.
The maximum number of amplicons in RNA designs is based on validation test results that demonstrated good gene expression dynamic range on a sequencing run. A good dynamic range means good sensitivity to detect extremes in the expression of genes in a given sample.
The last two columns of the IAD#_DataSheet.csv file (included in the download results files for an RNA design) reports the preferred TaqMan assay that can be used for verification of the particular target and classification of the TaqMan assay with 2 possible values:
• Classification 1: The recommended TaqMan Gene Expression Assay targets the same exon or exon-exon boundary as the Ion RNA AmpliSeq Design.
• Classification 2: The recommended TaqMan Gene Expression Assay targets the same set of RefSeq Accessions as the Ion RNA AmpliSeq Design.
Follow this link for instructions on how to get the corresponding TaqMan assay.
The high expressed genes were selected from rank-ordered lists of whole transcriptome RNA-Seq expression data derived from universal human reference RNA (UHRR, Stratagene). UHRR is comprised of purified RNAs from 10 distinct human cell lines and has been shown to be an accurate and reproducible standard for comparison of gene expression data. This reference RNA has been utilized by the highly referenced Microarray Quality Control (MAQC) consortium as well as the more recent Sequence Quality Control (SEQC) study. There is familiarity among microarrays users primarily as well as some RNA-Seq customers around the MAQC samples which also bolsters the decision to use this RNA for the rank ordered list.
Data Collection: Whole transcriptome sequence data was collected from both PGM and Proton runs using Ion 318 and Ion P1 chips, respectively.
Since UHRR represents several different tissues and we have a wealth of internal RNA-Seq data from this sample, we found UHRR to be a reasonable sample type for determining a common list of highly expressed genes.
References
While the depths of coverage on a 550 chip may be suitable for somatic variant calling with the Ion AmpliSeq exome panel, the recommendation for the 550 chip will be for germline variant calling only. Due to the 125-275 bp amplicon designs, we cannot guarantee the performance of the panel on degraded DNA from samples such as cfDNA or FFPE.
We are not currently considering this option, but its implementation will depend on the demand for it. Please contact your rep or our support team for product or feature requests.
Each Ion AmpliSeq Exome bundle (PN A38262 and A38264) has enough oligos for 8 reactions, or 8 exomes. are each provided with a 24 reaction Ion AmpliSeq Library kit Plus (PN 4488990) - which supports 8 exomes.
PN A38262 and A38264 have a shelf life of 15 months due to the library kit. The exome oligo plates are stable for 3 years. Please contact customer service for shelf life questions or COA documents.
On-target bases is the percentage of total sequenced bases that mapped to target regions. This metric reflects the percentage of bases from the amplicons that were designed, synthesized, and pooled that also generated sequence data that mapped back to the target regions.
The Ion AmpliSeq Designer takes into account many different parameters to compute the best set of amplicons to cover a target region. The ability to maximize in silico coverage depends upon factors such as repetitive regions and sequence complexity. The Ion AmpliSeq 2.0 User Guide provides guidance on how many amplicons can be combined to either the Ion 314 chip, the Ion 316 chip, or the Ion 318 chip. Our coverage uniform is >85% of amplicons is 0.2X of the mean coverage. If the mean (or average) coverage is 2000X, then 85% of the amplicons have a depth of coverage that is > 400X.
If you want additional coverage from your experiment, you could always run a larger chip or multiple chips. For example, you could simply take the same library that was constructed in your initial experiment, and then run another template prep and sequencing run on a subsequent chip in your second experiment. Currently, the recommendation to achieve ~500X coverage is to run ~1kb on an Ion 314 Chip, 50kb on an Ion 316 Chip, and 500kb on an Ion 318 Chip.
The AmpliSeq pipeline prepares alternative solutions that may have increased in-silico coverage over the default panel presented to the user as the recommended (default) result. Increased coverage is achieved by iteratively relaxing amplicon design constraints for amplicons overlapping the regions missed by the standard solutions. Although relaxing primer design constraints may result in increased off target amplification for these amplicons, this is limited to regions close to those missed by the default solution, and this approach had been demonstrated to be useful to many customers, while others may prefer the safe performance of the default High Specificity (recommended) solution.
No, you do not need to start all over. You can add, delete and edit genes or regions from the design results. You can even create a new version of the same design, with iterative modifications, and continue to resubmit the designs.
Variant confirmation can be done with other platforms, including Taqman SNP Genotyping Assays and Sanger sequencing–capillary sequencing.
If your design results in multiple pools and you are preparing libraries manually, then as per User Guide each pool should be amplified independently and amplification product shall be combined before primer digestion step for each sample.
Yes, you can pool your tumor and normal samples together into a single chip run (assuming that your target size and required coverage can be achieved in a single chip). Many people perform differential pooling so that the coverage of the normal sample is lower than the tumor sample.
We recommend using the Ion AmpliSeq Library Plus Kit as it is expected to produce higher library yield, increased uniformity and result in more robust library amplification than the standard Ion AmpliSeq 2.0 Library Kit.
There are a number of ways to perform orthogonal validation of mutations found by NGS, including TaqMan Genotyping Assays in standard or digital PCR formats, TaqMan Mutation Detection Assays for specific somatic mutations, or Sanger Sequencing using Capillary Electrophoresis.
To ensure that an entire exon is covered, by default we add 5 bp of padding up and down-stream of the selected target region to allow room to place the primers. Padding ensures that we are able to get good quality sequence at the ends of the exons and to get some sequence read into the splice junction regions. Primer regions are not considered covered. Therefore, if coverage obtained from the initial design is less than 100%, we can try once more to extend the primer further out into the intron to capture the whole exon.
Designs for pathogens are not currently supported for Ion AmpliSeq Designer.
It is possible to run 3 different AmpliSeq designs each with barcodes and combine them going into Template Prep.
Target regions from pre-designed PCR primer sets can certainly be imported and submitted for design into the Ion AmpliSeq Designer. The specific parameters of Ion AmpliSeq Designer may result in primer sets that are different from initially pre-designed PCR primer sets but are optimized for use with the Ion AmpliSeq Technology.
Due to quality control considerations after submission, amongst other properties, the .BED file is reviewed for duplicates. Once the duplicates have been removed and other filters have been passed, the (probably reduced) file is accepted into the pipeline.
The most common cause of a design request being marked as "Undesignable" is that AmpliSeq Designer is unable to generate any amplicons overlapping the target regions which satisfy the minimum design requirements.
Possible reasons for this include repeated sequences (e.g. pseudo-genes); low-complexity regions; very high or very low GC content or other problem sequences. These factors may prevent AmpliSeq Designer from being able to find pairs of primer locations which meet the minimum criteria for specificity, primer melting temperature, GC content, and so on. In such cases, we recommend checking the target sequences using a tool such as the UCSC genome browser. When using a custom reference, it is recommended that the sequence is manually checked by the user for the presence of duplicated or other problematic regions that might cause design requests to be undesignable. Note that, when uploading a custom reference, if the user selects an associated organism for specificity checks, the associated organism reference genome will be used in searching for repeats.
In addition, when using the Copy Amplicons functionality, AmpliSeq Designer will mark a design request as "Undesignable" if it is not able to fit all the copied amplicons into the available pools due to amplicon conflicts. Note that, while for AmpliSeq Custom designs, two amplicons conflict only if they overlap by more than the maximum allowable number of bases, in AmpliSeq HD some non-overlapping amplicons may also be flagged as conflicting due to other limitations of the AmpliSeq HD chemistry. In this situation, it may be necessary to remove some of the copied amplicons from the design.
An access code is used as part of the gateway for communication between AmpliSeq Designer, and the Ion Torrent analysis software, which can be either Torrent Suite or Ion Reporter.
If the user loses their Access Code, the recommendation is to generate a new one.
Yes, as long as the Access Code fulfills the minimum requirements, a custom Access Code is accepted.
Follow below instructions
This could be because of maintenance activity on thermofisher.com. Please wait until thermofisher.com website is back or check for the maintenance message on AmpliSeq.com for downtime notification.
This issue might be related to missing user profile details that are required for the AmpliSeq order workflow. Please contact regional AmpliSeq Support Americas , EMEA, Greater China, South Asia , Japan for assistance.
Cookies are blocked for AmpliSeq.com. Please enable cookies in the browser settings for successfully logging into the application.
Users with accounts assigned to a Supply Center are meant, by design, to use that account only for that purpose. Therefore, it is expected that they do not have access to other applications (including ampliseq.com) when using that type of account. Users who could log into ampliseq.com before, once their account is associated with a Supply Center cannot anymore login directly to ampliseq.com. However, in most cases, their ampliseq.com account can still be accessed by redirection from the Supply Center landing page (after logging in to thermofisher.com) by doing the following:
If the above doesn’t work and for users whose account has been directly setup as a Supply Center account, the only option to access ampliseq.com is to create a new account on thermofisher.com associated to a different email and not set up as a Supply Center account.