Publication Number MAN0010907 - Ion AmpliSeq Designer 7.4: Getting Started  (5.9 MB PDF download)
(5.9 MB PDF download)
Create and manage Reference Genomes
In AmpliSeq Designer, you can use a variety of human, animal, and plant reference genomes to build your panels. You can also upload your own. The steps below describe uploading your own.
- From the navigation bar, select Browse Pre-loaded Genomes.
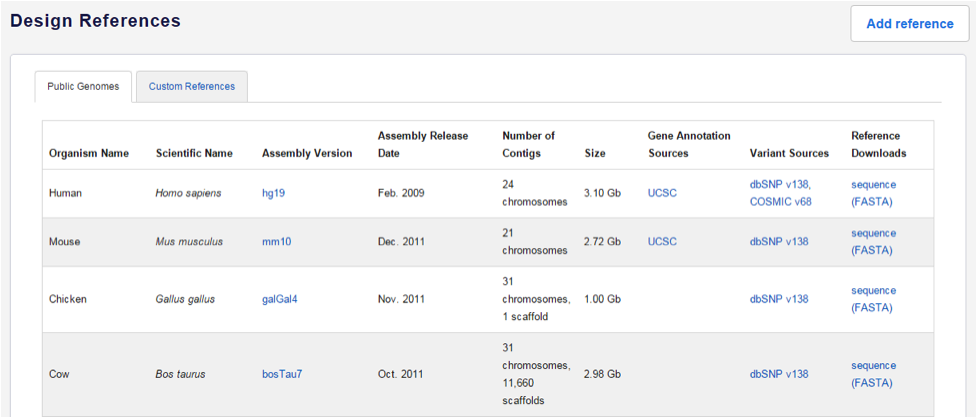
- To upload a new custom reference, click the Add reference button.
- Fill in the required information on the following screen:

- **Reference name**: Must be composed of US-ASCII letters, numbers, and spaces, between 3 and 32 characters in length.
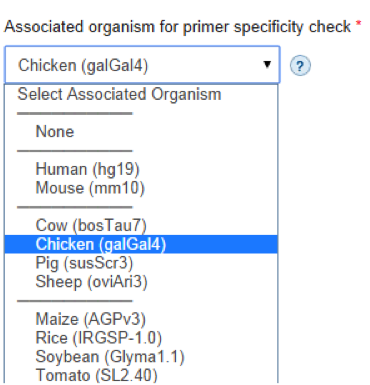
- **Associated organism for primer specificity check**: Click to view the dropdown menu containing list of organisms. If your data are associated with one of our supported organisms, providing this information may improve primer specificity to your custom reference by favoring primers with few optimal binding sites in the consensus sequence. Primer specificity check refers to the process of identifying potential primer mispriming events. Primers with high number of potential mispriming events are avoided in our designs.
- **Reference source (Recommended)**: Name the database/source of the DNA sequence.
- **Reference description**: Add any notes about the custom reference sequence.
- **Reference sequences**: You may either upload a FASTA file (Default size is 2.0 GB; however, upon request the limit can be extended to 4.0 GB):
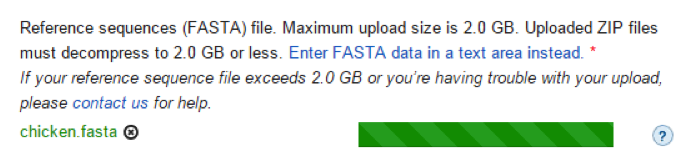 *or* copy and paste the reference sequence:
*or* copy and paste the reference sequence: 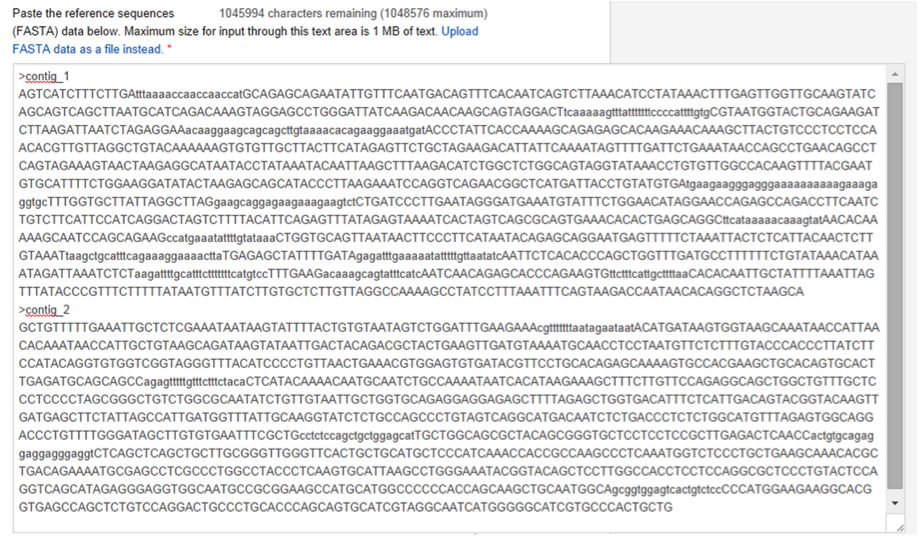
- **Genome short name for Torrent Server**: Should be composed of lowercase US-ASCII letters, numbers, and underscores, between 1 and 30 characters in length.
- **Known polymorphism (BED) file**: Indicates regions of the sequences in the custom reference FASTA file with high polymorphism (i.e., SNPs, indels, or other variation). Ion AmpliSeq Designer minimizes primer overlap with these regions. This file is optional. See Appendix for specifications on creating and formatting BED files for uploading.

- Save your custom reference by clicking the blue Save button and click on the Custom References tab to view your uploaded reference:
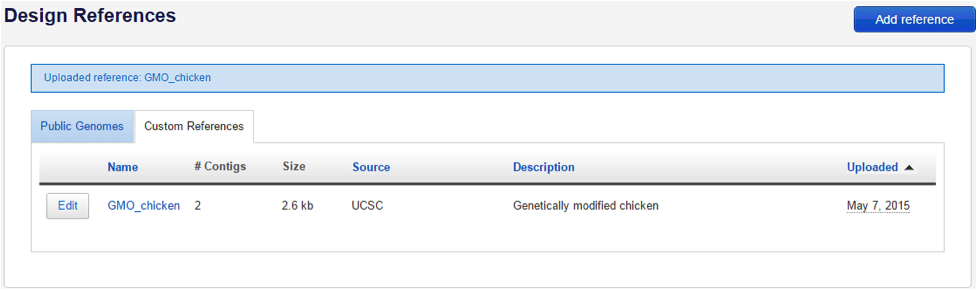
- Click on the custom reference name to show more information:
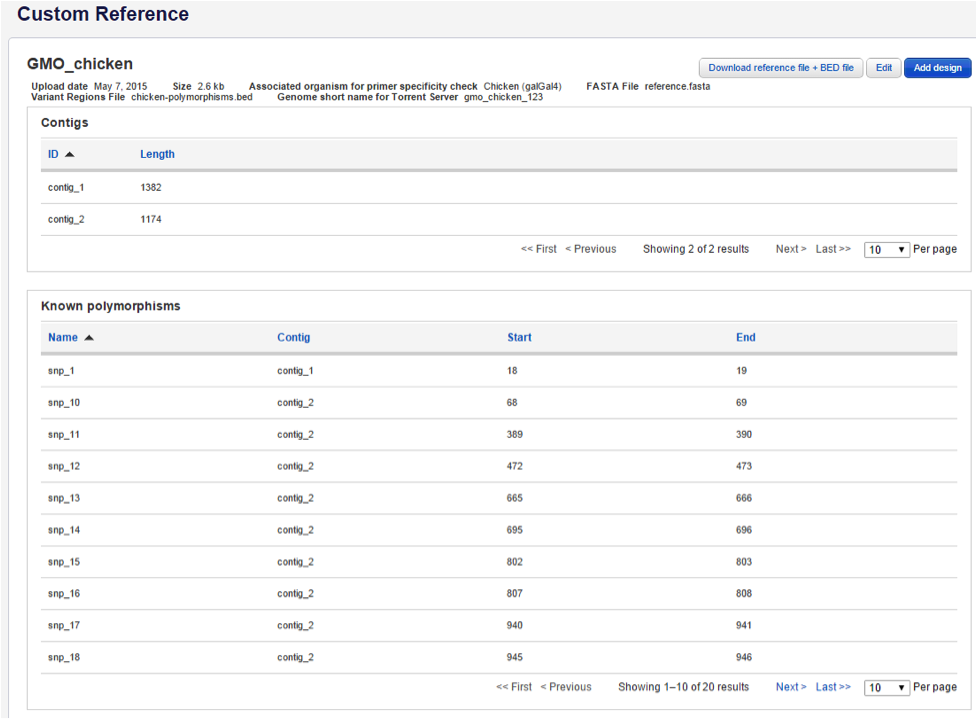
- Click on the blue Edit button in the upper right of the window to edit the following:
- Reference name
- Reference source
- Reference description
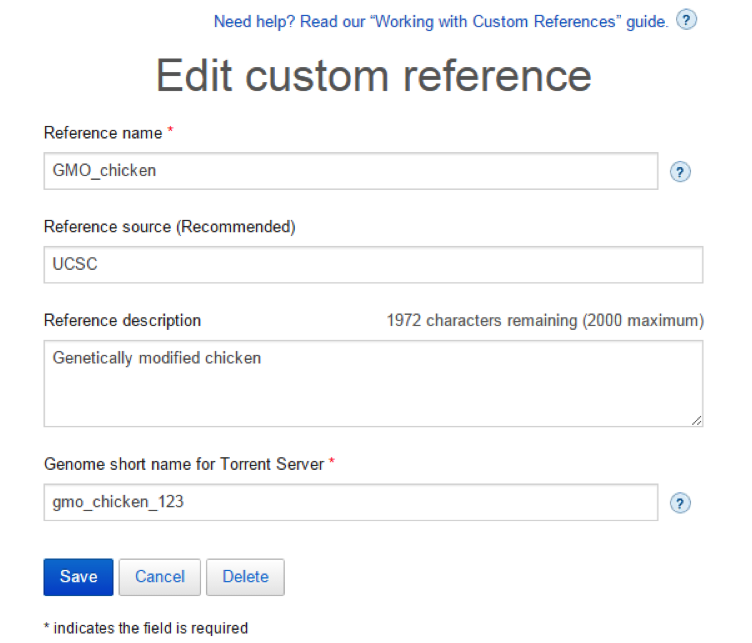
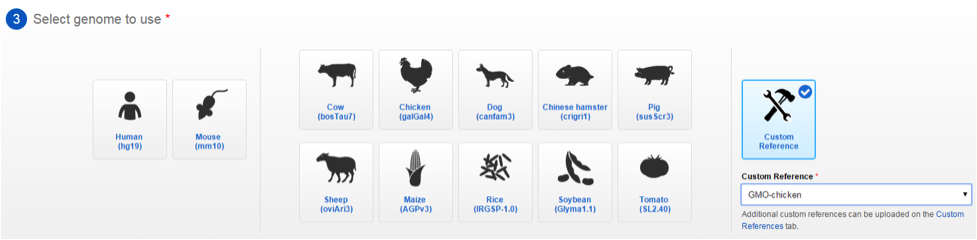
Note: You cannot make changes to the uploaded files (genomic data) as they are permanently associated with this assigned custom reference genome. If you want to make changes, you will need to delete and re-upload your edited files using the Add reference button. Click Delete to remove the reference from the list of active custom references. This will not affect existing designs; associated custom references will still be downloadable. 7. When building your custom panel, click Custom Reference and select your custom reference from the drop-down box.
Reference FASTA sequence
Uploading sequences
One or more reference sequences in FASTA format can be uploaded by:
- Selecting a "plain text" or compressed file (in either ZIP or GZIP formats) containing the sequence(s). The maximum file size allowed for upload is 2 GB uncompressed file.
- Copying and pasting the sequences in the text area available after clicking on the link "Enter FASTA data in a text area instead."
FASTA format
A sequence in FASTA format is expressed in 2 or more lines of text: The first line is an identifying "header", the rest of the lines (one or more) represent the sequence itself. NCBI downloaded fasta files are supported for upload for the following extensions: .fasta, .fa, .fasta.zip, .fasta.gz, .fna, .fna.gz (Follow the link to access the NCBI reference genomes).
- The header: The header line starts with a "greater-than" symbol (">") followed by at most 200 ASCII characters. Allowed characters are "A" to "Z", "a" to "z", "0" to "9", "_", "-", ".", ",", ";" and "|" with SPACES between them. Since the header is used to identify the sequence, it is required to be unique for each sequence in the reference.
- The sequence: The only characters accepted for representing a sequence are "A", "C", "G", "T" and "N" (lower case versions are also allowed for representing low complexity regions).The sequence of a contig can be described in a single or multiple lines after the header. If described by multiple lines, the sizes of each line has to be the same, except for the last line which can be smaller or longer than the previous lines. It is customary to use separate lines of 50 or 60 characters in length for readability reasons, when defining a contig sequence. For cases where a large single sequence line is used, the maximum size should not exceed 65535 bases. Any sequence exceeding this length as a single line, will trigger an error and is unsupported.
- Sequence size: The minimum length of a sequence is 160 bp: allowing 60 bp for minimum insert size, plus 50 bp upstream and 50 bp downstream to serve as a design buffer for primer positioning during amplicon design; however, the recommended upstream and downstream context buffer sequence for optimal designs is 1,000 bp.
Known polymorphism BED file
The known polymorphism BED file indicates regions of the sequences in the custom reference FASTA file with high polymorphism (i.e., SNPs, indels, or other variation). AmpliSeq Designer will minimize primer overlap with these regions. This file is optional. You may upload it at the time of creating a new custom reference.
The BED format is a tab-delimited file, with one line per region. Required fields are chrom, chromStart, and chromEnd in the first three columns of the BED file format. Additional fields will be ignored. The chrom field must match one contig ID in the accompanying FASTA file. chromStart and chromEnd fields are the zero-based, half-open coordinates indicating the region to target in the sequence identified by the ID in the chrom field. chromStart and chromEnd are relative to the sequence of the FASTA record corresponding to the given ID.
They must meet the following criteria:
- chromStart may be a value between 0 and length of the sequence specified by chrom minus 1.
- chromEnd must be greater than chromEnd.
- chromEnd may have a maximum value of the length of the sequence specified by chrom.
- No region should overlap any other region in the file. Overlapping regions should be merged by the customer into a single contiguous region.
Example FASTA file (50 bases per line):

Sample variants of interest (highlighted in blue):
| chrom |
chromStart |
chromEnd |
| contig_1 |
0 |
1 |
| contig_1 |
95 |
96 |
| contig_1 |
105 |
106 |
| contig_1 |
199 |
200 |
Sample formatted BED file:
contig_1 0 1
contig_1 95 96
contig_1 105 106
contig_1 199 200






